All in the Family
Published on
Children's DoctorPublished on
Children's DoctorRead a case study from the Cancer Center about a 15-year-old girl, previously healthy, whose family shares a history of cancer.
Kristin Zelley, MS, CGC, and Kim E. Nichols, MD
CL is a previously healthy 15-year-old girl who presents with a 2-week history of left-sided knee pain and progressive swelling. There is no history of fever or trauma. Physical examination reveals swelling of the left knee, but no redness, warmth, tenderness to palpation, or limitation in range of motion. A left knee X-ray reveals a lytic lesion in the distal femur, later confirmed by MRI. The mass is biopsied and found to be an osteosarcoma.
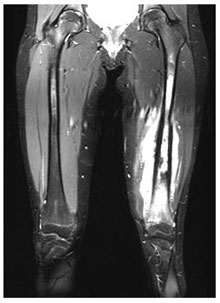 Photo: MRI of the left knee demonstrates an enhancing lesion of the distal femur with edema of the surrounding soft tissues.
Photo: MRI of the left knee demonstrates an enhancing lesion of the distal femur with edema of the surrounding soft tissues.
Upon questioning, CL’s mother notes that her husband died recently from acute myeloid leukemia following treatment for rhabdomyosarcoma. His sister has a history of breast cancer. Based on the family history of cancer, it was questioned whether CL might have Li-Fraumeni syndrome (LFS). She and her mother were counseled, and subsequently CL chose to undergo genetic testing. Test results returned positive for a mutation in TP53, the causative gene in LFS.
Discussion: CL has LFS, a rare cancer predisposing condition first described by Frederick Pei Li, MD, MA, and Joseph Fraumeni, MD, MSc, in 1969. Families with LFS are at greatest risk to develop six tumor types, including sarcomas of the muscle or bone, breast cancer, brain tumors, adrenocortical tumors, and acute leukemia. Since the original description, recent reports suggest that there are excess rates of many other cancer types. LFS is a highly penetrant syndrome, with more than 90% of affected individuals developing cancer by age 80. There is a significant risk for tumor development in childhood, with up to 20% of patients developing cancer during the first 2 decades of life. Affected individuals are at increased risk to develop multiple primary tumors, as in the case of CL’s father, and may be at increased risk for radiation- or therapy-induced cancers.
CL had a highly suspicious family cancer history, which is a strong indicator of a possible underlying predisposition syndrome (see Table 1). Family histories concerning for hereditary predisposition to cancer typically include: 1) multiple members on the same side of the family with the same or similar types of cancer, or distinct cancer types that group together in specific syndromes (such as the rhabdomyosarcoma, osteosarcoma, leukemia and breast cancer in this case); 2) relatives with early onset, multiple or bilateral cancers; and 3) a pattern suggestive of autosomal dominant inheritance. Therefore, when gathering a family history, clinicians should collect information on the age of cancer onset, type of cancer, and laterality in at least first- (parents and siblings), second- (grandparents, aunts, and uncles), and third- (cousins and great-grandparents) degree relatives. They should consider referral to a genetic counselor, oncologist, or geneticist with expertise in cancer predisposition if any of the features described below are identified.
It is important to recognize a hereditary cancer syndrome because identification of affected children allows for appropriate surveillance and cancer management. In pre-symptomatic children, the primary goal of surveillance is to detect cancers at the earliest stages, when they are small, more easily resected and require treatment with less intensive or even no chemotherapy. As a result, the chances for cure are maximized and risks of side effects reduced. In certain cases, children may also undergo prophylactic removal of at-risk organs, a procedure that eliminates or significantly lowers the lifetime risk for cancer. For those children already affected with cancer, genetic information can guide the choice of surgical approach or medical regimen. Finally, identification of affected children enables the testing of other family members who may or may not require similar surveillance, preventive, or treatment measures.
At CHOP, the Cancer Predisposition Program evaluates and manages children who have or are suspected of having a cancer-predisposing genetic condition. A pediatric oncologist, geneticist, genetic counselor, and psychologist work to address the needs of children who are at increased risk for cancer. Our team provides comprehensive medical management, recommends and reviews the results of screening tests, and offers supportive counseling. Each member of our team is dedicated to providing the most skilled, compassionate care available in an environment that focuses on children and their families. Also key to the program are community physicians, who are often the most likely to spot signs of predisposition in families, particularly among siblings and other relatives.
Strahm B, Malkin D. Hereditary cancer predisposition in children: genetic basis and clinical implications. Int J Cancer. 2006;119(9):2001-2006.
Rao A, Rothman JE, Nichols KE. Genetic testing and tumor surveillance for children with cancer predisposition syndromes. Curr Opin Pediatr. 2008;20(1):1-7.
Teplick A, Kowalski M, Biegel JA, Nichols KE. Screening in cancer predisposition syndromes: guidelines for the general pediatrician. Eur J Pediatr. 2011;170(3):285-94.
Knapke S, Zelley K, Nichols KE, Kohlman W, Schiffman J. Considerations for the identification, management, and genetic evaluation of children with cancer predisposing conditions. 2012. Am So Clin Oncol Educ Program.
Categories: Oncology, Children's Doctor Winter 2013