What is choanal atresia?
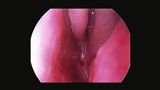
Choanal atresia is a congenital narrowing of the back of the nasal cavity that causes difficulty breathing. It is rare, occurring in approximately 1 in 7,000 live births, and is seen more often in females than in males. Choanal atresia is often associated with other developmental anomalies such as CHARGE, Treacher Collins syndrome, and Tessier syndrome.
If your child is diagnosed with choanal atresia, it may take two different forms. It can can be bilateral, meaning it blocks both nasal passages, or unilateral, meaning only one nasal passage is blocked. The blockage can be either completely bony (30 percent of cases) or be composed of both bone and membranes (70 percent of cases).
- Bilateral choanal atresia is life-threatening and symptoms appear immediately after birth. Babies breathe only through their noses when they are very young, so the blocked nasal passages will cause extreme difficulty breathing.
- Unilateral choanal atresia is more common, less serious, and sometimes appears later in childhood because the child has been able to manage while breathing through only one side of the nasal passage.
Causes
Choanal atresia is a developmental abnormality. The anomaly is present at birth and can be associated with other developmental abnormalities. There is no known specific cause of choanal atresia. Most believe that choanal atresia occurs when the tissue that separates the nose and mouth area during fetal development remains after birth.
Signs and symptoms
The symptoms of choanal atresia include:
- Cyclic respiratory distress relieved with crying
- Noisy breathing
- Feeding difficulties
- Inability to pass a 5/6 French suction catheter into the nasopharynx
- Nasal drainage
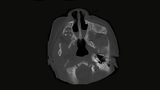
Further evaluation is done with endoscopy and CT scans.
Testing and diagnosis
Patients with bilateral choanal atresia often have respiratory distress episodes immediately after birth. These are relieved only when the patient begins to cry. The child can not breathe during normal circumstances and can only breath when they open their airway to cry. Because of this, infants with bilateral choanal atresia often require intubation immediately after delivery. Once the airway is secured, the patient can be evaluated for other developmental abnormalities.
Patients with unilateral choanal atresia often show symptoms later during infancy and childhood with signs of unilateral nasal obstruction (discharge, mouth breathing, inability to move air on one side). Since one side is open at birth, it is rare that these children have difficulty breathing when they are first born.
Aero-digestive evaluation
If your infant has choanal atresia, he should be evaluated for both airway and feeding issues. Management of the airway often requires a combination of supportive, medical and surgical care. Feeding and swallowing issues are very common in children with these conditions and often need to be addressed by speech pathologists and GI specialists. Children born with syndromes often need other subspecialty evaluations (cardiology, ophthalmology, etc.) and benefit greatly from the coordinated care provided in the multidisciplinary setting of the Center for Pediatric Airway Disorders.
Treatment
Treatment depends on the severity of the anomaly (unilateral or bilateral), how much it affects the child's breathing and eating, and what other medical conditions the patient has.
Treatment for mild cases
Sometimes, a child with unilateral choanal atresia has no respiratory distress or significant feeding issues. These patients may initially be managed with close observation and occasionally supplemental oxygen. Nasal saline can help to keep the nasal linings healthy and free of discharge. Parents with children diagnosed with unilateral choanal atresia may wait until the child grows older before deciding to repair the condition.
Treatment for severe cases
If your child is diagnosed with bilateral choanal atresia the condition must be treated as soon as possible. Surgery should be performed as soon as the patient is stable and has been evaluated for other anomalies. The major goal of the surgical repair is to open the nasal airway sufficiently, allowing the infant to breathe on his own . There are several approaches available to accomplish this goal, all of which are performed in the operating room under general anesthesia.
- Transnasal endoscopic approach: Currently, CHOP surgeons prefer to treat children with choanal atresia using the transnasal endoscopic approach. It can be done safely on infants just a few days old. The posterior nasal airway is opened using a combination of dilators, sinus instruments, ear curettes and drills. In some cases, a piece of endotracheal tube is placed inside the nasal passage as a stent to maintain the opening while the area heals. The stent will be removed a few weeks after the surgery at the bedside or during an office visit. Sometimes stenting is not required.
- Transpalatal approach: In rare circumstances where the skull base is abnormally developed (other craniofacial anomalies are present), the surgeon may need to use a transpalatal approach to reach the area of choanal atresia through the roof of the mouth.
What to know before your appointment
Prepare for your child's upcoming appointment with the Center for Pediatric Airway Disorders.
Resources to help
Center for Pediatric Airway Disorders Resources
We have created resources to help you find answers to your questions about airway disorders and feel confident with the care you are providing your child.

Meet your team
Our coordinated approach to care, bridging many specialties at CHOP, makes it possible to meet the complex needs of children who come to us for care.