
Five-year-old “Peter” presented to Clinical Genetics following evaluations by 27 subspecialists who were unable to provide his 2 physician parents with a unifying diagnosis to explain his seemingly disparate constellation of findings. But Peter’s parents were relentless in attempting to resolve their son’s diagnostic odyssey.
Peter was born at 38 weeks gestation following a pregnancy notable for IUGR. Neonatally, he had hyperbilirubinemia thrombocytopenia, and hypoglycemia. He was noted to have GERD, a submucosal cleft palate, dysphagia, constipation, and difficulty latching leading to failure to thrive. Later, he was diagnosed with growth hormone deficiency. At 4 months of age, he underwent bilateral inguinal hernia repair. Peter had chronic infections, including recurrent otitis media with resultant conductive hearing loss. Developmental milestones were delayed, particularly speech. An Orthopaedics assessment for hammer toes, flat feet, and patellar subluxation resulted in a negative connective tissue consult. No genomic testing was performed at that time, but an echocardiogram revealed a mildly dilated aortic root. At CHOP, the genetics dysmorphology exam was notable for malar flatness, hooded eyelids, protuberant ears, a bulbous nasal tip with hypoplastic alae nasi, micrognathia, and long, slender fingers.
A microarray was sent, and a few weeks later revealed a chromosome 22q11.2 deletion—the most common cause of DiGeorge syndrome (DGS), despite his lack of classic triad of DGS: congenital heart disease, hypocalcemia, and immunodeficiency. Although comforted in having a diagnosis of 22q11.2 deletion syndrome (22q11.2DS), the parents voiced concerns regarding the possibility of having missed treatable associated features earlier.
Diagnosis leads to recognition of additional symptoms
Following the diagnosis, Peter was found to have growth hormone deficiency, idiopathic scoliosis, recurrent patellar subluxation, ADHD, anxiety, insomnia, and a nonverbal learning disability with strengths in reading and rote memorization and deficits in mathematics and abstract reasoning. His submucosal cleft palate required surgical repair, addressing his nasopharyngeal reflux and hypernasal speech.
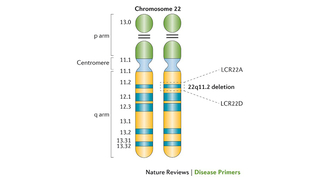
22q11.2 deletion syndrome is the most common microdeletion, identified in 1 in 992 pregnancies and 1 in 2,148 live births, resulting in loss of ~50 genes leading to significant morbidity and some mortality. The first description of these constellation of findings was in the 1960s in children with DGS. Throughout the 1980s, unbalanced translocations involving chromosome 22 in patients with features of DGS were reported, including initially by CHOP’s Elaine H. Zackai, MD, and Beverly S. Emanuel, PhD, leading to identification of cytogenetically visible chromosome 22q11.2 deletions in a subset of patients with DGS. But what about the remaining patients with DGS where no chromosome 22q11.2 deletion was visible under the microscope? This led to the development of fluorescence in situ hybridization (FISH) probes specific to chromosome 22q11.2 in the United Kingdom and at CHOP in the early 1990s, identifying a chromosome 22q11.2 deletion (see Figure 1) in most patients with DGS. This revolutionized diagnostic capabilities, establishing the high prevalence of the condition.
Following the introduction of FISH studies, 22q11.2DS was found in additional diagnoses with overlapping features, including velocardiofacial (VCFS), conotruncal anomaly face (CTAF), and Cayler cardiofacial syndromes, as well as in some patients with Opitz G/BBB syndrome. Importantly, the 22q11.2 deletions observed in these patients were identical, providing indisputable evidence for a common etiology across all conditions. Today, the term DGS is generally reserved for those rare instances when the etiology is not caused by a 22q11.2 deletion, eg, in association with CHARGE syndrome (due to CHD7 mutations on chromosome 8) and teratogens, such as retinoic acid or diabetic embryopathy. Otherwise, the broad phenotypic spectrum, with or without features of classic DGS, is referred to by the cytogenetic nomenclature— 22q11.2DS, providing a unifying diagnosis for families, caregivers, payors, and support organizations alike.
Multifaceted, variable, developing condition
22q11.2DS is quite variable and can present with any combination of congenital anomalies, medical issues, cognitive deficits, behavioral differences, and later onset conditions (see Figure 2)—all far extending the original description of DGS. Thus, management requires a lifetime multidisciplinary approach with coordinated care and genetic counseling, as although >90% of 22q11.2 deletions arise as a de novo event in the child, once present, a person with the deletion has a 50% chance of having a child with 22q11.2DS.

Importantly, 22q11.2DS is common. It is the most common cause of syndromic palatal abnormalities and schizophrenia, and the second most common cause of congenital heart disease and developmental delay after Down syndrome. The presence of associated features varies by age and the focus changes over time, with an emphasis on managing congenital anomalies, particularly congenital heart disease, associated medical issues, such as recurrent infection, hypocalcemia, feeding and swallowing difficulties, delays in acquisition of motor milestones and language, in early childhood; optimizing speech and addressing cognitive challenges in early school age; and shifting attention to behavioral differences, independence, later onset conditions, and the 50% recurrence risk in adolescences and adulthood.
Peter’s case shows that, although common, lack of recognition of associated features and/or lack of familiarity with genetic testing methods—together with such wide variability—often delays diagnosis. Conversely, early identification has the potential to improve outcomes. Equally important, 22q11.2DS has become a model for understanding rare and frequent congenital anomalies, medical conditions, psychiatric illness, and cognitive deficits, providing opportunities for translational strategies across the lifespan for both patients with 22q11.2DS and individuals with associated features in the general population.
CHOP’s 22q and You Center is considered the world leader in clinical care and research initiatives for chromosome 22q differences. It is the largest such center in the United States and draws patients from all over the country and the world, giving it unparalleled expertise and experience diagnosing and caring for individuals with 22q11.2DS.
References and further readings:
Campbell IM, et al. (2018). What is new with 22q? An update from the 22q and You Center at the Children’s Hospital of Philadelphia. AM J Med Genet. Oct 31. 176 (10):2058-2069.
Bassett AS, McDonald-McGinn DM, et al. Practical guidelines for managing patients with 22q11.2 deletion syndrome. J Pediatr. 2011;159(2):332-339.
McDonald-McGinn, DM. 22q11.2 deletion — a tiny piece leading to a big picture. Nat Rev Dis Primers. 2020;6(1):33.
Featured in this article
Specialties & Programs
Five-year-old “Peter” presented to Clinical Genetics following evaluations by 27 subspecialists who were unable to provide his 2 physician parents with a unifying diagnosis to explain his seemingly disparate constellation of findings. But Peter’s parents were relentless in attempting to resolve their son’s diagnostic odyssey.
Peter was born at 38 weeks gestation following a pregnancy notable for IUGR. Neonatally, he had hyperbilirubinemia thrombocytopenia, and hypoglycemia. He was noted to have GERD, a submucosal cleft palate, dysphagia, constipation, and difficulty latching leading to failure to thrive. Later, he was diagnosed with growth hormone deficiency. At 4 months of age, he underwent bilateral inguinal hernia repair. Peter had chronic infections, including recurrent otitis media with resultant conductive hearing loss. Developmental milestones were delayed, particularly speech. An Orthopaedics assessment for hammer toes, flat feet, and patellar subluxation resulted in a negative connective tissue consult. No genomic testing was performed at that time, but an echocardiogram revealed a mildly dilated aortic root. At CHOP, the genetics dysmorphology exam was notable for malar flatness, hooded eyelids, protuberant ears, a bulbous nasal tip with hypoplastic alae nasi, micrognathia, and long, slender fingers.
A microarray was sent, and a few weeks later revealed a chromosome 22q11.2 deletion—the most common cause of DiGeorge syndrome (DGS), despite his lack of classic triad of DGS: congenital heart disease, hypocalcemia, and immunodeficiency. Although comforted in having a diagnosis of 22q11.2 deletion syndrome (22q11.2DS), the parents voiced concerns regarding the possibility of having missed treatable associated features earlier.
Diagnosis leads to recognition of additional symptoms
Following the diagnosis, Peter was found to have growth hormone deficiency, idiopathic scoliosis, recurrent patellar subluxation, ADHD, anxiety, insomnia, and a nonverbal learning disability with strengths in reading and rote memorization and deficits in mathematics and abstract reasoning. His submucosal cleft palate required surgical repair, addressing his nasopharyngeal reflux and hypernasal speech.
22q11.2 deletion syndrome is the most common microdeletion, identified in 1 in 992 pregnancies and 1 in 2,148 live births, resulting in loss of ~50 genes leading to significant morbidity and some mortality. The first description of these constellation of findings was in the 1960s in children with DGS. Throughout the 1980s, unbalanced translocations involving chromosome 22 in patients with features of DGS were reported, including initially by CHOP’s Elaine H. Zackai, MD, and Beverly S. Emanuel, PhD, leading to identification of cytogenetically visible chromosome 22q11.2 deletions in a subset of patients with DGS. But what about the remaining patients with DGS where no chromosome 22q11.2 deletion was visible under the microscope? This led to the development of fluorescence in situ hybridization (FISH) probes specific to chromosome 22q11.2 in the United Kingdom and at CHOP in the early 1990s, identifying a chromosome 22q11.2 deletion (see Figure 1) in most patients with DGS. This revolutionized diagnostic capabilities, establishing the high prevalence of the condition.
Following the introduction of FISH studies, 22q11.2DS was found in additional diagnoses with overlapping features, including velocardiofacial (VCFS), conotruncal anomaly face (CTAF), and Cayler cardiofacial syndromes, as well as in some patients with Opitz G/BBB syndrome. Importantly, the 22q11.2 deletions observed in these patients were identical, providing indisputable evidence for a common etiology across all conditions. Today, the term DGS is generally reserved for those rare instances when the etiology is not caused by a 22q11.2 deletion, eg, in association with CHARGE syndrome (due to CHD7 mutations on chromosome 8) and teratogens, such as retinoic acid or diabetic embryopathy. Otherwise, the broad phenotypic spectrum, with or without features of classic DGS, is referred to by the cytogenetic nomenclature— 22q11.2DS, providing a unifying diagnosis for families, caregivers, payors, and support organizations alike.
Multifaceted, variable, developing condition
22q11.2DS is quite variable and can present with any combination of congenital anomalies, medical issues, cognitive deficits, behavioral differences, and later onset conditions (see Figure 2)—all far extending the original description of DGS. Thus, management requires a lifetime multidisciplinary approach with coordinated care and genetic counseling, as although >90% of 22q11.2 deletions arise as a de novo event in the child, once present, a person with the deletion has a 50% chance of having a child with 22q11.2DS.
Importantly, 22q11.2DS is common. It is the most common cause of syndromic palatal abnormalities and schizophrenia, and the second most common cause of congenital heart disease and developmental delay after Down syndrome. The presence of associated features varies by age and the focus changes over time, with an emphasis on managing congenital anomalies, particularly congenital heart disease, associated medical issues, such as recurrent infection, hypocalcemia, feeding and swallowing difficulties, delays in acquisition of motor milestones and language, in early childhood; optimizing speech and addressing cognitive challenges in early school age; and shifting attention to behavioral differences, independence, later onset conditions, and the 50% recurrence risk in adolescences and adulthood.
Peter’s case shows that, although common, lack of recognition of associated features and/or lack of familiarity with genetic testing methods—together with such wide variability—often delays diagnosis. Conversely, early identification has the potential to improve outcomes. Equally important, 22q11.2DS has become a model for understanding rare and frequent congenital anomalies, medical conditions, psychiatric illness, and cognitive deficits, providing opportunities for translational strategies across the lifespan for both patients with 22q11.2DS and individuals with associated features in the general population.
CHOP’s 22q and You Center is considered the world leader in clinical care and research initiatives for chromosome 22q differences. It is the largest such center in the United States and draws patients from all over the country and the world, giving it unparalleled expertise and experience diagnosing and caring for individuals with 22q11.2DS.
References and further readings:
Campbell IM, et al. (2018). What is new with 22q? An update from the 22q and You Center at the Children’s Hospital of Philadelphia. AM J Med Genet. Oct 31. 176 (10):2058-2069.
Bassett AS, McDonald-McGinn DM, et al. Practical guidelines for managing patients with 22q11.2 deletion syndrome. J Pediatr. 2011;159(2):332-339.
McDonald-McGinn, DM. 22q11.2 deletion — a tiny piece leading to a big picture. Nat Rev Dis Primers. 2020;6(1):33.
Contact us
22q and You Center