

Case:
Baby girl L.M. was born at 37 weeks, 1 day via c-section to a G1P1 mother. Cell-free DNA testing performed prenatally had previously revealed a 46,XX karyotype, so L.M.’s parents were surprised when their baby girl was born with ambiguous genitalia resembling a phallic like structure. Her genitalia were graded as a 5 on the Prader Scale. (See Figure 1.)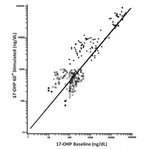
Her newborn screening sent on day 1 of life revealed a 17-alpha-hydroxyprogesterone (17OHP) level of 932 ng/mL (presumptive positive >30 ng/mL) and a 17OHP extracted of 200 ng/mL (presumptive positive >19 ng/mL), raising suspicion for congenital adrenal hyperplasia (CAH), and she came under the care of CHOP’s Adrenal and Puberty Center. A confirmatory 17OHP level drawn on day 2 of life was 13,291 ng/dL, a critical high value. Additional adrenal androgens, including androstenedione and testosterone, were also markedly elevated. This prompted her team to perform a high-dose ACTH stimulation test. Results were interpreted using the ACTH stimulation test nomogram, and L.M. plotted in the range of classical congenital adrenal hyperplasia. Her baseline 17OHP was 14429 ng/dL and her a-60 minute stimulated 17OHP rose to 24,201 ng/dL. (See Figure 2.)
The genetics team was then consulted for CYP21A2 gene sequencing, and L.M. was found to be a homozygote for a pathogenic variant defined as c.293-13C>G (aka In2G), which typically correlates with salt-wasting CAH. A postnatal karyotype was sent and confirmed 46,XX female. A pelvic ultrasound revealed a normal uterus, although her right and left ovaries were not visualized.
Discussion: L.M. was then started on replacement therapy with hydrocortisone, fludrocortisone, and NaCl tablets. She was initially started on a moderate stress dose of hydrocortisone (~25 mg/m2/day) for about 2 weeks and then weaned down to physiologic hydrocortisone given 3 times a day, with her highest dose of hydrocortisone being given at bedtime to suppress the overnight rise of ACTH. She was given a stress dose steroid plan, and the family received training in the Connelly Family Learning Center prior to discharge. Her parents learned various situations during which stress dosing would be required, as well as how to administer an intramuscular injection of hydrocortisone sodium succinate (such as Solu-Cortef®).
Parents were instructed to have a medic-alert bracelet made that says “Adrenal Insufficiency” or “Steroid Dependent,” rather than “congenital adrenal hyperplasia” so emergency responders are able to quickly recognize the needs of the child. L.M. was given a referral to follow up with Urology at around age 3 months of life; her family is interested in learning more about vaginoplasty options and timing, although they are aware that this surgery is not absolutely mandatory should they choose to defer until a later date.
Ongoing Monitoring and Management
This is an example of a typical case of classical CAH diagnosed immediately after birth. Following diagnosis, children with CAH are typically followed in outpatient Endocrinology every 3 months until they reach adulthood. Standard laboratory monitoring includes adrenal androgens, plasma renin activity, and electrolytes every 3 months; labs are checked first thing in the morning as this is when hormone values spike in the blood. Medication doses are titrated according to hormone values. As the child grows, they will continue to require increasing doses of medication to match their increasing body surface area. Typically once a child reaches age 2 or 3 years and can regulate their sodium intake through their diet, then their NaCl tablets may be discontinued. As a child reaches school age, monitoring bone-age X-rays and watching for signs of precocious puberty will also become standard practice at Endocrinology visits. Children with CAH are at higher risk for developing precocious puberty and, should that occur, they can be treated with a gonadotropin-releasing hormone agonist (GnRHa) such as leuprolide acetate (Lupron®) or histrelin acetate (Supprelin®).
Children with CAH do not have any activity restrictions; they are able to attend school, play sports, and go on to live long, healthy, and productive lives. As they enter adulthood and are considering starting families, we will advise them to meet with a genetic fertility counselor to understand the potential risk of passing the autosomal recessive disorder to their children.
When caring for a child with CAH, it is important to know these children are not immune to the usual ailments of childhood, including ear infections, rashes, and broken bones, and therefore should be managed accordingly. In addition to standard treatments, they will also require stress dose steroids during times of fever, vomiting, surgery, etc. Each child will have an individualized stress dose steroid plan (usually given as oral hydrocortisone) but they will also have IM Solu-Cortef in case of emergency.
In addition to CAH, CHOP’s Adrenal and Puberty Center specializes in caring for children and adolescents with adrenal insufficiency, delayed and precocious puberty, hypogonadism and other related disorders. The center’s interdisciplinary team includes endocrinologists, urologists, geneticists, and fertility specialists and has unique clinics for patients with Klinefelter syndrome and disorders of sexual development. It is active in research and is currently enrolling in 2 industry studies that are testing new therapies for CAH.
Reference and Suggested Reading
- Congenital Adrenal Hyperplasia Guideline Resources. Endocrine Society Web site. Accessed September 25, 2019.
Featured in this article
Specialties & Programs
Case:
Baby girl L.M. was born at 37 weeks, 1 day via c-section to a G1P1 mother. Cell-free DNA testing performed prenatally had previously revealed a 46,XX karyotype, so L.M.’s parents were surprised when their baby girl was born with ambiguous genitalia resembling a phallic like structure. Her genitalia were graded as a 5 on the Prader Scale. (See Figure 1.)
Her newborn screening sent on day 1 of life revealed a 17-alpha-hydroxyprogesterone (17OHP) level of 932 ng/mL (presumptive positive >30 ng/mL) and a 17OHP extracted of 200 ng/mL (presumptive positive >19 ng/mL), raising suspicion for congenital adrenal hyperplasia (CAH), and she came under the care of CHOP’s Adrenal and Puberty Center. A confirmatory 17OHP level drawn on day 2 of life was 13,291 ng/dL, a critical high value. Additional adrenal androgens, including androstenedione and testosterone, were also markedly elevated. This prompted her team to perform a high-dose ACTH stimulation test. Results were interpreted using the ACTH stimulation test nomogram, and L.M. plotted in the range of classical congenital adrenal hyperplasia. Her baseline 17OHP was 14429 ng/dL and her a-60 minute stimulated 17OHP rose to 24,201 ng/dL. (See Figure 2.)
The genetics team was then consulted for CYP21A2 gene sequencing, and L.M. was found to be a homozygote for a pathogenic variant defined as c.293-13C>G (aka In2G), which typically correlates with salt-wasting CAH. A postnatal karyotype was sent and confirmed 46,XX female. A pelvic ultrasound revealed a normal uterus, although her right and left ovaries were not visualized.
Discussion: L.M. was then started on replacement therapy with hydrocortisone, fludrocortisone, and NaCl tablets. She was initially started on a moderate stress dose of hydrocortisone (~25 mg/m2/day) for about 2 weeks and then weaned down to physiologic hydrocortisone given 3 times a day, with her highest dose of hydrocortisone being given at bedtime to suppress the overnight rise of ACTH. She was given a stress dose steroid plan, and the family received training in the Connelly Family Learning Center prior to discharge. Her parents learned various situations during which stress dosing would be required, as well as how to administer an intramuscular injection of hydrocortisone sodium succinate (such as Solu-Cortef®).
Parents were instructed to have a medic-alert bracelet made that says “Adrenal Insufficiency” or “Steroid Dependent,” rather than “congenital adrenal hyperplasia” so emergency responders are able to quickly recognize the needs of the child. L.M. was given a referral to follow up with Urology at around age 3 months of life; her family is interested in learning more about vaginoplasty options and timing, although they are aware that this surgery is not absolutely mandatory should they choose to defer until a later date.
Ongoing Monitoring and Management
This is an example of a typical case of classical CAH diagnosed immediately after birth. Following diagnosis, children with CAH are typically followed in outpatient Endocrinology every 3 months until they reach adulthood. Standard laboratory monitoring includes adrenal androgens, plasma renin activity, and electrolytes every 3 months; labs are checked first thing in the morning as this is when hormone values spike in the blood. Medication doses are titrated according to hormone values. As the child grows, they will continue to require increasing doses of medication to match their increasing body surface area. Typically once a child reaches age 2 or 3 years and can regulate their sodium intake through their diet, then their NaCl tablets may be discontinued. As a child reaches school age, monitoring bone-age X-rays and watching for signs of precocious puberty will also become standard practice at Endocrinology visits. Children with CAH are at higher risk for developing precocious puberty and, should that occur, they can be treated with a gonadotropin-releasing hormone agonist (GnRHa) such as leuprolide acetate (Lupron®) or histrelin acetate (Supprelin®).
Children with CAH do not have any activity restrictions; they are able to attend school, play sports, and go on to live long, healthy, and productive lives. As they enter adulthood and are considering starting families, we will advise them to meet with a genetic fertility counselor to understand the potential risk of passing the autosomal recessive disorder to their children.
When caring for a child with CAH, it is important to know these children are not immune to the usual ailments of childhood, including ear infections, rashes, and broken bones, and therefore should be managed accordingly. In addition to standard treatments, they will also require stress dose steroids during times of fever, vomiting, surgery, etc. Each child will have an individualized stress dose steroid plan (usually given as oral hydrocortisone) but they will also have IM Solu-Cortef in case of emergency.
In addition to CAH, CHOP’s Adrenal and Puberty Center specializes in caring for children and adolescents with adrenal insufficiency, delayed and precocious puberty, hypogonadism and other related disorders. The center’s interdisciplinary team includes endocrinologists, urologists, geneticists, and fertility specialists and has unique clinics for patients with Klinefelter syndrome and disorders of sexual development. It is active in research and is currently enrolling in 2 industry studies that are testing new therapies for CAH.
Reference and Suggested Reading
- Congenital Adrenal Hyperplasia Guideline Resources. Endocrine Society Web site. Accessed September 25, 2019.
Contact us
Adrenal and Puberty Center