About CCAM
Congenital cystic adenomatoid malformation is a benign malformation of the lung that is usually diagnosed during the mid-gestational screening ultrasound as a “bright” lesion in the lung tissue that may or may not contain visible cysts. During prenatal life, CCAMs may grow rapidly until 25 to 28 weeks' gestation, when they usually plateau in size, and then usually decrease in size relative to the fetus during the remainder of gestation.
Although a very large CCAM may cause heart failure (hydrops) in the fetus and require fetal intervention, the majority of CCAMs are asymptomatic at birth. In postnatal life, CCAMs usually present with infection, typically as difficult-to-treat pneumonias that are resistant to antibiotic treatment. They may also rupture a cyst, and present with lung collapse (pneumothorax). Finally, CCAMs have been documented to turn into malignant tumors over time. For these reasons, we recommend early resection of even asymptomatic CCAMs, typically prior to 2 months of age.
Case study
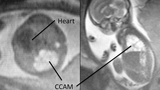
A 32-year-old woman was referred to CHOP at 23 weeks' gestation for a cystic lung mass detected on routine screening obstetrical ultrasound examination at 21 weeks' gestation. An ultrasound and MRI (figure 1) confirmed the presence of a right-sided, multicystic lung mass, with compression of the fetal heart to the left side and no other anomalies. She was followed by serial ultrasound assessment for the remainder of her pregnancy, during which the mass decreased in size relative to the fetus and the heart returned to a normal position. She was counseled that the infant would be asymptomatic at birth, and it was recommended that she deliver at a hospital close to home and that she return to CHOP for postnatal imaging of the lesion and treatment recommendations after 1 month of age.
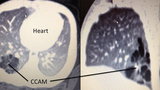
As anticipated, the infant was asymptomatic after a normal vaginal delivery and continued to thrive at home until he returned to CHOP at 5 weeks of age. A CT angiogram study at that time confirmed the persistence of a right lower lobe multicystic CCAM (figure 2), and it was recommended that the baby undergo a thoracoscopic right lower lobectomy. At 6 weeks of age, the baby was taken to the operating room and underwent successful resection of the lesion using thoracoscopic technique (figure 3).
Thoracoscopic lobectomy to treat CCAM
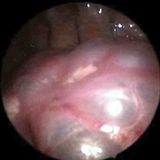
Thoracoscopic lobectomy in infants is an advanced, minimally invasive procedure that is performed through three 5mm trocars passed through the chest wall. High definition visualization and magnification is provided by a 4mm diameter thoracoscope and the dissection and ligation of the blood vessels and bronchus (airway) to the lobe is accomplished using small instruments passed through the two instrument ports.
Thoracoscopic surgery offers the advantages of less pain and a potentially better long-term functional and cosmetic result.
The infant was taken to our Harriet and Ronald Lassin Newborn/Infant Intensive Care Unit after surgery and was discharged home in normal condition on the second postoperative day.
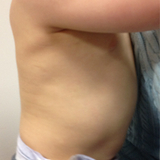
At one year of follow-up, he has healed with minimal scar formation and is developing normally (figure 4).
Featured in this article
Specialties & Programs
About CCAM
Congenital cystic adenomatoid malformation is a benign malformation of the lung that is usually diagnosed during the mid-gestational screening ultrasound as a “bright” lesion in the lung tissue that may or may not contain visible cysts. During prenatal life, CCAMs may grow rapidly until 25 to 28 weeks' gestation, when they usually plateau in size, and then usually decrease in size relative to the fetus during the remainder of gestation.
Although a very large CCAM may cause heart failure (hydrops) in the fetus and require fetal intervention, the majority of CCAMs are asymptomatic at birth. In postnatal life, CCAMs usually present with infection, typically as difficult-to-treat pneumonias that are resistant to antibiotic treatment. They may also rupture a cyst, and present with lung collapse (pneumothorax). Finally, CCAMs have been documented to turn into malignant tumors over time. For these reasons, we recommend early resection of even asymptomatic CCAMs, typically prior to 2 months of age.
Case study
A 32-year-old woman was referred to CHOP at 23 weeks' gestation for a cystic lung mass detected on routine screening obstetrical ultrasound examination at 21 weeks' gestation. An ultrasound and MRI (figure 1) confirmed the presence of a right-sided, multicystic lung mass, with compression of the fetal heart to the left side and no other anomalies. She was followed by serial ultrasound assessment for the remainder of her pregnancy, during which the mass decreased in size relative to the fetus and the heart returned to a normal position. She was counseled that the infant would be asymptomatic at birth, and it was recommended that she deliver at a hospital close to home and that she return to CHOP for postnatal imaging of the lesion and treatment recommendations after 1 month of age.
As anticipated, the infant was asymptomatic after a normal vaginal delivery and continued to thrive at home until he returned to CHOP at 5 weeks of age. A CT angiogram study at that time confirmed the persistence of a right lower lobe multicystic CCAM (figure 2), and it was recommended that the baby undergo a thoracoscopic right lower lobectomy. At 6 weeks of age, the baby was taken to the operating room and underwent successful resection of the lesion using thoracoscopic technique (figure 3).
Thoracoscopic lobectomy to treat CCAM
Thoracoscopic lobectomy in infants is an advanced, minimally invasive procedure that is performed through three 5mm trocars passed through the chest wall. High definition visualization and magnification is provided by a 4mm diameter thoracoscope and the dissection and ligation of the blood vessels and bronchus (airway) to the lobe is accomplished using small instruments passed through the two instrument ports.
Thoracoscopic surgery offers the advantages of less pain and a potentially better long-term functional and cosmetic result.
The infant was taken to our Harriet and Ronald Lassin Newborn/Infant Intensive Care Unit after surgery and was discharged home in normal condition on the second postoperative day.
At one year of follow-up, he has healed with minimal scar formation and is developing normally (figure 4).
Contact us
Richard D. Wood Jr. Center for Fetal Diagnosis and Treatment