High-Risk Neuroblastoma: Using Proton Therapy as Part of Multi-modality Treatment
Published on
Children's DoctorPublished on
Children's DoctorRead a case study from the Cancer Center about a 3-year-old girl presenting with difficulty sleeping, increased crying, and abdominal pain, diagnosed with neuroblastoma.
Christine Hill-Kayser, MD, and Naomi Balamuth, MD
A 3-year-old girl presents with difficulty sleeping, increased crying, and abdominal pain. She is the product of a normal full-term delivery and has no significant past medical history. Her parents note that she was healthy until approximately a month ago, when she began waking at night complaining of pain. They also note that she has been increasingly irritable and less willing to walk over the past 2 to 3 weeks. They deny fever, vomiting, diarrhea, and constipation. She has not lost any weight that they have noticed, although her appetite has been dwindling.
On examination, the patient is in no acute distress, although appears somewhat quiet and clingy. An abdominal mass is palpable in the region above the umbilicus. Her lungs are clear, and the remainder of her exam is within normal limits. CT scan of the chest, abdomen, and pelvis shows a large mass in the central abdomen, extending cranially to the mid-thorax along the right paraspinous region. Bone marrow biopsy demonstrates small round blue cells. Urine catecholamines are somewhat elevated above normal.
Discussion: The diagnosis is neuroblastoma, a malignancy that arises from embryonic neural crest cells of the peripheral nervous system and is the most common extra-cranial solid tumor of childhood. The presentation and stage at diagnosis, as well as the associated prognosis, may be very heterogeneous. Several diagnostic elements factor into risk stratification, including patient age, disease stage, tumor cell ploidy, histopathology, MYCN gene copy number, and other genetic aspects of the tumor. Select patients who are very young (less than 1 year of age) may have disease designated stage 4S that is expected to spontaneously regress and have an excellent prognosis. Patients with lower intermediate-risk (stage 1 or 2) disease have an excellent prognosis after treatment with surgical resection with or without chemotherapy. Unfortunately, however, about half of patients have high-risk disease (stage 3 or 4, depending on other diagnostic factors) at diagnosis. Although the prognosis for such children is guarded, with aggressive treatment, long-term survival rates approach 50 percent. The current treatment paradigm for children with high-risk neuroblastoma is intensive multi-agent induction chemotherapy followed by surgical resection of the primary tumor, high-dose chemotherapy followed by autologous stem cell transplant (ASCT), radiation therapy, and immunotherapy.
The patient described above was ultimately diagnosed with high-risk neuroblastoma. She underwent induction chemotherapy that included topotecan, cyclophosphamide, cisplatin, etoposide, vincristine, and doxorubicin. Surgical resection of her abdominothoracic mass was undertaken; the abdominal component was completely resected although the chest mass could not be entirely removed.
Following ASCT, the patient received radiation therapy. Historically, X-ray therapy has been used to deliver this treatment for neuroblastoma patients; in most centers worldwide, either 3D conformal X-ray therapy or intensity-modulated X-ray therapy would have been employed. Proton therapy is a radiation technology that has been available since the 1970s and has become more widespread over the past 1 to 2 decades. Proton therapy differs from X-ray therapy in that protons can be made to stop within tissue. Compared to X-rays, which traverse the patient depositing both entrance and exit dose, protons, with their ability to target smaller areas, allow considerable sparing of normal tissues. Proton therapy for neuroblastoma is a relatively new approach.
For this patient, the use of proton therapy compared to X-ray therapy allowed a100-fold reduction in radiation dose delivered to the patient’s normal liver, lungs, and heart (see Figure 1). This was particularly important as it reduced her risk of pulmonary toxicity that was increased previously by her chemotherapy. It also reduced her risk of liver and kidney toxicity, allowing her to quickly receive the needed immunotherapy following completion of her radiation. The patient tolerated radiation with no clinical toxicity and is currently disease-free.
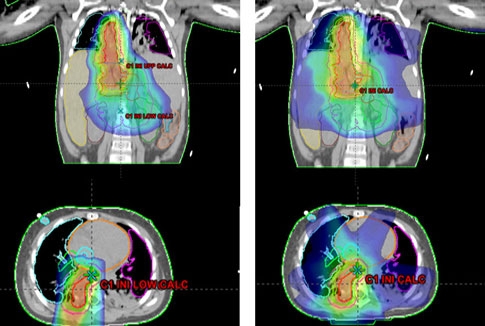
Figure 1: Radiation treatment plans for delivery of radiation to the tumor bed in the abdomen and chest using protons (left) versus x-ray therapy (right). Note the relative sparing of the liver and lungs when proton therapy is employed.
Proton therapy shows great promise for patients such as the one described here, and is available to many patients with solid and brain tumors who travel from around the world to receive treatment at The Children’s Hospital of Philadelphia/Roberts Proton Therapy Center.
Yu AL, Gilman AL, Ozkaynak MF, et al; Children’s Oncology Group. Anti-GD2 antibody with GM-CSF, interleukin-2, and isotretinoin for neuroblastoma. N Engl J Med. 2010;363(14):1324-1334.
Hill-Kayser CE, Both S, Tochner Z. Proton therapy: ever shifting sands and the opportunities and obligations within. Front Oncol. 2011;1:24.
Hattangadi JA, Rombi B, Yock TI, et al. Proton radiotherapy for high-risk pediatric neuroblastoma: early outcomes and dose comparison. Int J Radiat Oncol Biol Phys. 2012;83(3):1015-1022.
For physician-to-physician questions and guidance regarding pediatric cancer, call CHOP’s Physician-Only line at 1-888-ONC-CHOP (1-888 662-2467) or email choprefonco@email.chop.edu.
To reach the specific programs highlighted in this issue, call 215-615-5678 for Proton Therapy; 267-426-5877 for the Pediatric Hereditary Cancer Predisposition Program; and 215-590-0432 for the Cancer Survivorship Program.
Categories: Oncology, Children's Doctor Winter 2013