What is non-syndromic craniosynostosis?
Craniosynostosis is a condition in which the sutures (growth seams) in an infant’s skull close too early, causing problems with normal brain and skull growth. Non-syndromic craniosynostosis is a non-inherited, isolated finding without related anomalies such as disorders of the limbs, ears or cardiovascular system. It typically involves the early closure of a single growth seam, or suture, in your child’s skull.
The specific head shape, appearance and diagnosis of a patient with non-syndromic craniosynostosis depends on what suture is affected. The most common form of non-syndromic craniosynostosis involves the sagittal suture. Other common forms include coronal, metopic and lambdoidal sutures.
Sagittal synostosis
Also known as: scaphocephaly
Suture involved: sagittal suture, the seam that runs from the front to the back of the skull
Cause and occurrence: the most common form of non-syndromic craniosynostosis, it typically occurs in prenatal development
Clinical characteristics:
- Head shape that is long and narrow, growing forward and backward due to restriction growing side to side
- Forehead may appear larger and the sides of the skull appear narrow

Unicoronal synostosis
Also known as: anterior plagiocephaly, unilateral coronal synostosis
Suture involved: one of the coronal sutures, the seams that runs from the ear to the soft spot on top of the baby’s head
Cause and occurrence: the cause is unknown but can be related to underlying associated genetic malformation in upwards of 20% to 30% of cases; it is more frequently found on the left side of the head.
Clinical characteristics:
- Asymmetrical forehead and brow
- Lack of forward growth on the affected side, resulting in flatness and retrusion
- Harlequin deformity: the eye socket on the affected side appears taller than the other eye, resulting in an eye that appears more widely open, and the bridge of the nose is bent toward the affected side of the face

The right eyebrow is raised with retrusive forehead and the nasal root is deviated to the right in this instance of unicoronal synostosis.
Bicoronal synostosis
Also known as: brachycephaly
Suture(s) involved: both of the coronal sutures, the seams that run from ear to ear over top of the head and meet at the soft spot in the middle
Cause and occurrence: closure of these sutures prevents normal forward growth of the skull, forcing the head to grow from side to side
Clinical characteristics:
- Disproportionately wide and flat head
- Flat back of the head and often extreme forehead incline (height)

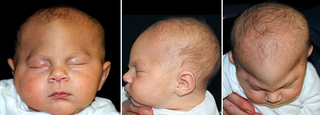
Metopic synostosis
Also known as: trigonocephaly
Suture involved: metopic suture, the seam that runs from the top of the head down the middle of the forehead to the nose
Cause and occurrence: early closure of the suture during prenatal development; generally an isolated condition. In rare cases, however, the presence of hypotelorism (close-set eyes) in trigonocephaly can be associated with other congenital anomalies. Of note: the metopic suture closes normally around 6 to 8 months of age. If closure happens slightly early, there may be a small ridge of the forehead, known as a metopic ridge, without further changes to the shape of the skull or evidence of trigonocephaly. Treatment is conservative observation.
Clinical characteristics:
- Triangular-shaped forehead when viewed from above
- Eyes may appear closer together
- Forehead will appear to jut out in the middle or appear more prominent when looking directly at the face
Lambdoid synostosis
Also known as: posterior plagiocephaly
Suture involved: one or both of the lambdoid sutures, the two seams located on the back of the skull
Cause and occurrence: very rare and the least common form of non-syndromic craniosynostosis
Clinical characteristics:
- Trapezoid shape
- Early closure causes a flattening on the back of the head on the same side where the suture has fused
- On the affected side, the ear is often posteriorly displaced, moved toward the back of the head or set lower than the non-affected ear, which is a distinguishing feature
- Bossing, or protrusion, at the back of the head on the non-affected side
- Atypical bulge in the mastoid region generally visible

Above, left: Asymmetric flattening of the back lower-right side of the head, characteristic of unilateral lambdoid synostosis. Right: Growth restriction on the back-right side of the head with compensatory growth of the brain and skull to the left.
Depiction of the typical characteristics of lambdoid synostosis. Note the flatness on the back-right side with bossing (protrusion) on the back left. The right ear is displaced posteriorly (backwards).

Positional plagiocephaly
Also known as: deformational plagiocephaly
Cause and occurrence: head shape change that is NOT related to premature closure of a suture, typically related to environmental factors or sleeping position. Deformational plagiocephaly does not cause a restriction of brain growth and usually remodels over the first few years of an infant’s life.
Clinical characteristics:
- Parallelogram shape
- Flatness on the back of the affected side of the skull with compensatory prominence of the forehead on the same side
- Ear displacement forward on the affected (same) side
- Non-affected side on the back of the skull protrudes with compensatory retrusion of the forehead on the same side
Treatment options:
- Frequent position changes during the day
- Adjustment of crib orientation in the nursery or car seat to encourage looking to the opposite side
- Limit time spent in car seats
- Tummy time while awake and supervised
- Physical therapy to treat torticollis (head tilt due to neck muscle problems), if present, as this limits neck range of motion and mobility
- Cranial molding helmet therapy in moderate to severe cases, typically initiated around 6 months of age
Complex multiple suture synostosis
Suture(s) involved: multiple sutures in the baby’s skull, creating a more serious condition than any other form of single suture synostosis; can cause a number of skull deformities
Cause and occurrence: the cause may be sporadic or genetic, though more likely to be associated with a genetic syndrome such as Apert, Crouzon or Pfeiffer syndromes; could indicate underlying brain abnormalities such as hydrocephalus or Arnold-Chiari malformation
Minor suture synostosis
Suture(s) involved: minor sutures are extensions of major sutures around the base of the skull
Cause and occurrence: rare; progressive head shape abnormality in the setting of patent (open) major cranial sutures (sagittal, coronal, metopic and lambdoid); surgery may be needed if there is a concern for constriction affecting brain growth or development of elevated intracranial pressure
Clinical characteristics:
- Skull asymmetry in variable ways
- Persistent or worsening skull deformity
Diagnosis
Each of these conditions requires a consultation with clinicians who specialize in the treatment of children with craniofacial anomalies. A thorough physical exam may initially diagnose the condition. Confirmation of the diagnosis is done with a 3D head CT scan. The CT scan allows your child’s medical team to closely evaluate atypical findings and rule out any associated anomalies. This is an important step in establishing the best treatment plan if the abnormal head shape is related to craniosynostosis.
Treatment
Treatment of single suture or multi-suture non-syndromic craniosynostosis depends on the suture involved, the extent of the abnormality, and the age of the child. If left untreated, 10% to 15% of patients with a single suture affected may go on to develop elevated intracranial pressure, thus requiring surgery. The craniofacial abnormalities may also result in severe physical differences.
Surgical treatment of non-syndromic craniosynostosis
CHOP’s Craniofacial Program offers the full spectrum of treatment options for patients with non-syndromic craniosynostosis. Your child’s care will be managed by the nation’s top pediatric plastic and reconstructive surgeons working closely with pediatric neurosurgery specialists.
Depending on the age of your child, the type of suture involved, and the degree of deformity, a variety of interventions may be recommended. Learn more about the current surgical treatment approach for children with craniosynostosis »
Non-surgical treatment options
In mild cases of craniosynostosis, surgery may not be required.
For example, in the case of slightly premature closure of the metopic suture with resulting mild metopic ridge and no other indication of trigonocephaly, treatment is typically conservative observation, as this will continue to change over time.
Watch this educational video to learn more about the treatment approaches available to children with different types of craniosynostosis.
Resources to help
Craniofacial Program Resources
We have gathered resources to give you information and help you find answers to your questions. We hope this makes your family's life a little easier.

Meet your team
Every person on your child’s team has the same goal: to see your child thrive. We provide medical care, emotional support and much more. Each team member has extensive experience in treating children with visible differences.
Reviewed by Scott P. Bartlett, MD, Jesse A. Taylor, MD