Case
A 24-month-old African-American boy presented to his pediatrician for a well-child check. He had no complaints and an unremarkable physical exam. Complete blood count sent as routine 2-year-old screening showed pancytopenia with hemoglobin 6.1 g/dL, a WBC count 3.6 K/uL, and platelet count of 120 K/uL. Differential was not initially available. Family history was unremarkable. Pennsylvania newborn screen had been normal for hemoglobin screening. The patient was referred to the Emergency Department, where he was afebrile with normal vital signs. On targeted questioning, parents noted decreased activity and appetite with increased fatigue. Recent viral symptoms were denied. Exam was notable for pallor and a 2/6 systolic ejection murmur, with no jaundice or hepatosplenomegaly. Repeat blood counts in the ED were essentially the same. White blood cell differential analysis showed 14% neutrophils, 84% lymphocytes, 2% atypical lymphocytes, and calculated absolute neutrophil count of 520 per uL. He received a transfusion of packed red blood cells and was discharged home with Hematology follow-up. Over the next 2 months, he required 2 pRBC transfusions for hemoglobin
Discussion
Sideroblasts are red blood cell precursor cells that have abnormal mitochondrial iron accumulation. While numerous causes of sideroblastic anemia exist, mitochondrial respiratory chain disease is among the most common. Mutations in more than 350 genes have been found to cause mitochondrial disease, which are energy deficiency disorders having highly variable signs and symptoms. There is no universal phenotype, biomarker, age of onset, or mode of inheritance. While patients may present with an apparently isolated symptom, a common hallmark of mitochondrial respiratory chain disease is progressive multisystem involvement over time that may only become apparent with focused evaluations. In this case, further testing revealed lactic acidemia and exocrine pancreatic insufficiency.
The co-occurrence of sideroblastic anemia and exocrine pancreatic insufficiency in a young child is a classical presentation for single large-scale mitochondrial DNA deletion syndrome (SLSMDS), classically labeled as Pearson syndrome. However, SLSMDS patients commonly present with diverse phenotypes and changing organ involvement that may evolve over a patient’s lifespan. (See figure 1)
Figure 1. Over time, the organs involved in single large-scale mitochondrial DNA deletion syndrome change.
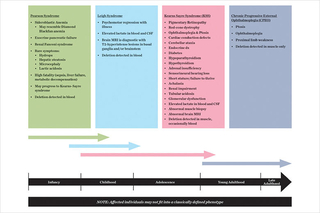
Patients do not always fit into the existing classical syndromic phenotypes for single large-scale mitochondrial DNA (mtDNA) deletions, but instead show a variety of features from among the subtypes of SLSMDS. All patients who are discovered to have SLSMDS should therefore be similarly screened and managed (see table 1 below).
SLSMDS is caused by a large mtDNA genome deletion (ranging from 1 to 10 kilobases, with a 5 kilobase deletion being common). mtDNA is comprised of 37 genes, all of which are essential for mitochondrial respiratory chain function to generate energy. Unlike nuclear genes, mtDNA is nearly always maternally inherited. A single large deletion occurring anywhere within the circular mtDNA genome causes SLSMDS, and when present, typically has arisen in a sporadic fashion rather than through maternal inheritance.
Multiple mtDNA deletions, instead of single deletions, typically indicate a patient has a mtDNA maintenance disorder caused by one of multiple different nuclear gene disorders. The optimal genetic diagnostic approach to identify mtDNA deletions is by whole mitochondrial DNA genome sequencing using next generation sequencing, often starting in blood. In patients without hematologic involvement, however, the mtDNA deletion may only be found in DNA isolated from muscle.
The Department of Pathology at Children’s Hospital of Philadelphia now offers mtDNA genome sequencing as a stand-alone clinical diagnostic test or performed together with whole exome sequencing. When whole exome sequencing is being performed for suspected mitochondrial disease, it is prudent to simultaneously order mtDNA genome sequencing. When mtDNA genome deletions are detected, their heteroplasmy level is quantified by a reflex digital droplet qPCR test performed in CHOP Pathology.
Genetic counseling and clinical management for children identified to have mitochondrial disease, including SLSMDS, is available in CHOP’s Mitochondrial Medicine Frontier Program.
Table 1: CHOP’s team to manage patients with single large-scale mitochondrial DNA deletion syndrome
Ophthalmologic
CHOP Mito specialists: Bart Leroy, MD; Grant Liu, MD; Robert Avery, DO, MSCE; Tomas Aleman, MD (Ophthalmology); William Katowitz, MD (Oculoplastics)
Clinical management:
- Yearly dilated exams, consider electroretinogram (ERG) and optical coherence tomography (OCT)
- Surgical ptosis repair
- Prism glasses for diplopia
Audiology
Clinical management:
- Yearly hearing screening
- Hearing aids; cochlear implants
Central nervous system
CHOP Mito specialists: Neurology/Neuromuscular
Clinical management:
- Measure CSF 5-MTHF (if low treat with folinic acid 3 mg/kg/day divided twice daily)
- Physical and occupational therapies
- Physical medicine and rehabilitation consult for assistive devices
Cardiac
CHOP Mito specialists: Anirban Banerjee, MD (Cardiology), Electrophysiology
Clinical management:
- Electrocardiogram (EKG) every 6 months to evaluate for cardiomyopathy and arrhythmias
- Consider pacemaker/defibrillator prophylactically, especially if WPW or conduction block seen
Endocrine
CHOP Mito specialists: Shana McCormack, MD, MTR
Clinical management:
- Annual screen for diabetes mellitus (HgbA1C, fasting glucose)
- Annual screen for adrenal insufficiency (8 am cortisol level, ACTH)
- Annual screen for hypoparathyroidism (calcium, PTH level)
- Annual screen for hypothyroidism (TSH, Free T4, T4)
- Growth and nutrition evaluation, and diet optimization
Hematologic
CHOP Mito specialists: Helge Hartung, MD
Clinical management:
- CBC/differential
- pRBC transfusions for anemia
Gastrointestinal
CHOP Mito specialists: Kristin Fiorino, MD; John Boyle, MD; Robert Heuckeroth, MD, PhD (Dysmotility Center); Kathleen Loomes, MD (Hepatology)
Clinical management:
- Measure fecal elastase — if abnormal consider pancreatic enzyme replacement
- Evaluate for GI dysmotility — if present, consider periactin or erythromycin
Renal
CHOP Mito specialists: Kevin Meyers, MBBCh
Clinical management:
- Monitor for renal tubular acidosis and glomerular dysfunction
- Consider sodium bicarbonate supplementation
- Monitor electrolytes and supplement as needed
References and suggested readings
DiMauro S, Hirano M. Mitochondrial DNA deletion syndromes. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle: University of Washington, Seattle; 2003 Dec 17 [Updated May 3, 2011]. 1993-2018.
Ducamp, S, Fleming, MD. The molecular genetics of sideroblastic anemia. [Published online ahead of print November 6, 2018.] Blood.
Parikh S, Goldstein A, Karaa A, et al. Patient care standards for primary mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genet Med. 2017;19(12).
Featured in this article
Specialties & Programs
Case
A 24-month-old African-American boy presented to his pediatrician for a well-child check. He had no complaints and an unremarkable physical exam. Complete blood count sent as routine 2-year-old screening showed pancytopenia with hemoglobin 6.1 g/dL, a WBC count 3.6 K/uL, and platelet count of 120 K/uL. Differential was not initially available. Family history was unremarkable. Pennsylvania newborn screen had been normal for hemoglobin screening. The patient was referred to the Emergency Department, where he was afebrile with normal vital signs. On targeted questioning, parents noted decreased activity and appetite with increased fatigue. Recent viral symptoms were denied. Exam was notable for pallor and a 2/6 systolic ejection murmur, with no jaundice or hepatosplenomegaly. Repeat blood counts in the ED were essentially the same. White blood cell differential analysis showed 14% neutrophils, 84% lymphocytes, 2% atypical lymphocytes, and calculated absolute neutrophil count of 520 per uL. He received a transfusion of packed red blood cells and was discharged home with Hematology follow-up. Over the next 2 months, he required 2 pRBC transfusions for hemoglobin
Discussion
Sideroblasts are red blood cell precursor cells that have abnormal mitochondrial iron accumulation. While numerous causes of sideroblastic anemia exist, mitochondrial respiratory chain disease is among the most common. Mutations in more than 350 genes have been found to cause mitochondrial disease, which are energy deficiency disorders having highly variable signs and symptoms. There is no universal phenotype, biomarker, age of onset, or mode of inheritance. While patients may present with an apparently isolated symptom, a common hallmark of mitochondrial respiratory chain disease is progressive multisystem involvement over time that may only become apparent with focused evaluations. In this case, further testing revealed lactic acidemia and exocrine pancreatic insufficiency.
The co-occurrence of sideroblastic anemia and exocrine pancreatic insufficiency in a young child is a classical presentation for single large-scale mitochondrial DNA deletion syndrome (SLSMDS), classically labeled as Pearson syndrome. However, SLSMDS patients commonly present with diverse phenotypes and changing organ involvement that may evolve over a patient’s lifespan. (See figure 1)
Figure 1. Over time, the organs involved in single large-scale mitochondrial DNA deletion syndrome change.
Patients do not always fit into the existing classical syndromic phenotypes for single large-scale mitochondrial DNA (mtDNA) deletions, but instead show a variety of features from among the subtypes of SLSMDS. All patients who are discovered to have SLSMDS should therefore be similarly screened and managed (see table 1 below).
SLSMDS is caused by a large mtDNA genome deletion (ranging from 1 to 10 kilobases, with a 5 kilobase deletion being common). mtDNA is comprised of 37 genes, all of which are essential for mitochondrial respiratory chain function to generate energy. Unlike nuclear genes, mtDNA is nearly always maternally inherited. A single large deletion occurring anywhere within the circular mtDNA genome causes SLSMDS, and when present, typically has arisen in a sporadic fashion rather than through maternal inheritance.
Multiple mtDNA deletions, instead of single deletions, typically indicate a patient has a mtDNA maintenance disorder caused by one of multiple different nuclear gene disorders. The optimal genetic diagnostic approach to identify mtDNA deletions is by whole mitochondrial DNA genome sequencing using next generation sequencing, often starting in blood. In patients without hematologic involvement, however, the mtDNA deletion may only be found in DNA isolated from muscle.
The Department of Pathology at Children’s Hospital of Philadelphia now offers mtDNA genome sequencing as a stand-alone clinical diagnostic test or performed together with whole exome sequencing. When whole exome sequencing is being performed for suspected mitochondrial disease, it is prudent to simultaneously order mtDNA genome sequencing. When mtDNA genome deletions are detected, their heteroplasmy level is quantified by a reflex digital droplet qPCR test performed in CHOP Pathology.
Genetic counseling and clinical management for children identified to have mitochondrial disease, including SLSMDS, is available in CHOP’s Mitochondrial Medicine Frontier Program.
Table 1: CHOP’s team to manage patients with single large-scale mitochondrial DNA deletion syndrome
Ophthalmologic
CHOP Mito specialists: Bart Leroy, MD; Grant Liu, MD; Robert Avery, DO, MSCE; Tomas Aleman, MD (Ophthalmology); William Katowitz, MD (Oculoplastics)
Clinical management:
- Yearly dilated exams, consider electroretinogram (ERG) and optical coherence tomography (OCT)
- Surgical ptosis repair
- Prism glasses for diplopia
Audiology
Clinical management:
- Yearly hearing screening
- Hearing aids; cochlear implants
Central nervous system
CHOP Mito specialists: Neurology/Neuromuscular
Clinical management:
- Measure CSF 5-MTHF (if low treat with folinic acid 3 mg/kg/day divided twice daily)
- Physical and occupational therapies
- Physical medicine and rehabilitation consult for assistive devices
Cardiac
CHOP Mito specialists: Anirban Banerjee, MD (Cardiology), Electrophysiology
Clinical management:
- Electrocardiogram (EKG) every 6 months to evaluate for cardiomyopathy and arrhythmias
- Consider pacemaker/defibrillator prophylactically, especially if WPW or conduction block seen
Endocrine
CHOP Mito specialists: Shana McCormack, MD, MTR
Clinical management:
- Annual screen for diabetes mellitus (HgbA1C, fasting glucose)
- Annual screen for adrenal insufficiency (8 am cortisol level, ACTH)
- Annual screen for hypoparathyroidism (calcium, PTH level)
- Annual screen for hypothyroidism (TSH, Free T4, T4)
- Growth and nutrition evaluation, and diet optimization
Hematologic
CHOP Mito specialists: Helge Hartung, MD
Clinical management:
- CBC/differential
- pRBC transfusions for anemia
Gastrointestinal
CHOP Mito specialists: Kristin Fiorino, MD; John Boyle, MD; Robert Heuckeroth, MD, PhD (Dysmotility Center); Kathleen Loomes, MD (Hepatology)
Clinical management:
- Measure fecal elastase — if abnormal consider pancreatic enzyme replacement
- Evaluate for GI dysmotility — if present, consider periactin or erythromycin
Renal
CHOP Mito specialists: Kevin Meyers, MBBCh
Clinical management:
- Monitor for renal tubular acidosis and glomerular dysfunction
- Consider sodium bicarbonate supplementation
- Monitor electrolytes and supplement as needed
References and suggested readings
DiMauro S, Hirano M. Mitochondrial DNA deletion syndromes. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle: University of Washington, Seattle; 2003 Dec 17 [Updated May 3, 2011]. 1993-2018.
Ducamp, S, Fleming, MD. The molecular genetics of sideroblastic anemia. [Published online ahead of print November 6, 2018.] Blood.
Parikh S, Goldstein A, Karaa A, et al. Patient care standards for primary mitochondrial disease: a consensus statement from the Mitochondrial Medicine Society. Genet Med. 2017;19(12).
Contact us
Mitochondrial Medicine Program